Pathophysiology
Among other physiological deficiencies, patients with type 2 diabetes are resistant to insulin. The liver and muscles develop resistance to insulin 5 to 10 years before most patients are diagnosed with diabetes, a condition known as pre-diabetes. Early in the course of the disease, pancreatic beta-cells are able to offset this resistance by secreting more insulin, thereby maintaining a normoglycemic environment.1 However, insulin resistance worsens over time and beta-cells begin to fail, leading initially to increases in post-prandial glucose levels. As insulin secretion declines further and the liver begins overproducing glucose at night, fasting plasma glucose levels subsequently rise.1,2 Over time, worsening glucotoxicity results in further insulin resistance, progressive beta-cell dysfunction, and loss of beta-cell mass. Once insulin secretion no longer keeps pace with insulin resistance, plasma glucose levels remain persistently elevated between 160 to 300 mg/dL.1 On average, at diagnosis patients have lost 50%-80% of their beta-cell function.2,3
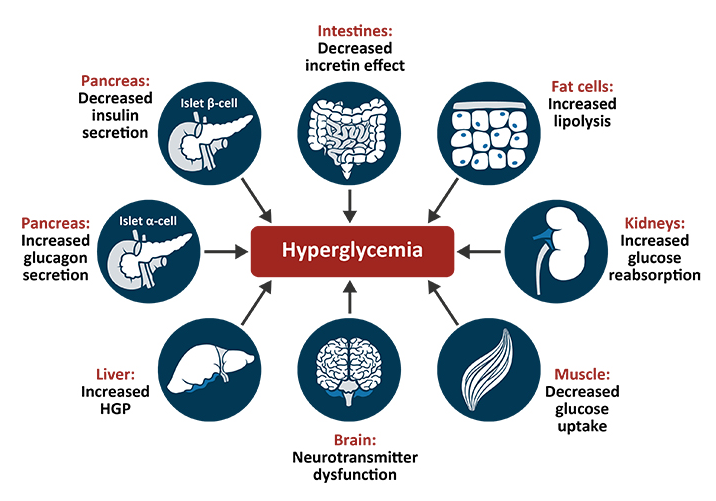
Beta-cell dysfunction is only one of at least 8 core physiological defects implicated in the pathogenesis of type 2 diabetes. Other sites of dysfunction include:
- Muscles
In the muscles, insulin resistance prevents the uptake of glucose after the ingestion of carbohydrates and reduces glycogen synthesis and storage, leading to postprandial hyperglycemia.2
- Liver
Insulin resistance in the liver is manifested by an increase in the rate of basal hepatic glucose production despite the presence of significantly elevated fasting insulin levels. The loss of response to insulin leads to fasting hyperglycemia and it is exacerbated by the increase in glucagon secretion from pancreatic alpha-cells.2
- Gastrointestinal system
In response to food, the gut releases two incretin hormones, GLP-1 and GIP. Incretin hormones bind to beta-cells and increase insulin secretion in a glucose-dependent manner. GLP-1 and GIP are responsible for up to 70% of insulin secretion following a meal. However, in patients with type 2 diabetes, the production of incretins is significantly reduced and beta-cells become resistant to the stimulatory effects of incretins on insulin secretion.4,5
- Pancreatic alpha-cells
In patients with diabetes, pancreatic alpha-cells oversecrete glucagon. After the ingestion of a meal, postprandial glucagon secretion is not suppressed but instead paradoxically increases in patients with type 2 diabetes. In addition, enhanced hepatic sensitivity to glucagon leads to increased basal hepatic glucose production, further contributing to hyperglycemia.2
- Brain
Central nervous system resistance to the effects of insulin contributes to a decreased release of neurotransmitters in the brain, leading to hunger, weight gain, and peripheral insulin resistance in muscles and liver.2
- Kidneys
In healthy individuals, filtered glucose is reabsorbed until the maximal reabsorptive capacity of the kidney is reached. If the concentration of glucose exceeds this capacity, excess glucose is excreted in urine to help maintain normal plasma glucose levels. In patients with type 2 diabetes, upregulation of glucose transporters increases glucose reabsorption by the kidneys, contributing to the development of hyperglycemia.2,6
- Adipocytes
Insulin is an antilipolytic hormone that blocks the breakdown of lipids. In insulin resistance, fat cells continually breakdown triglycerides into free fatty acids. Elevated intracellular levels of toxic lipid metabolites lead to insulin resistance in the liver and muscles, increased hepatic gluconeogenesis, decreased insulin secretion, and beta-cell failure and apoptosis.2
Some antidiabetic agents simultaneously target several pathophysiologic defects implicated in type 2 diabetes. For example, GLP-1 receptor agonists mimic the incretin hormone GLP-1 and reduce hyperglycemia by stimulating insulin production from beta-cells only in the presence of elevated glucose levels. As these agents stimulate insulin release only when blood glucose concentrations are above fasting levels, the actions of GLP-1 receptor agonists are self-terminating and unlikely to induce hypoglycemia.5 Additionally, GLP-1 receptor agonists also improve metabolic control by inhibiting glucagon secretion, slowing gastric emptying and the absorption of glucose, and suppressing appetite.7 Some evidence suggests that GLP-1 receptor agonists may also promote beta-cell proliferation and reduce beta-cell apoptosis. The regeneration of beta cells may improve glucose homeostasis over time as it may compensate for the loss of beta-cell function observed in type 2 diabetes.8,9 Currently approved GLP-1 receptor agonists include exenatide, semaglutide, liraglutide, lixisenatide, and dulaglutide.
The pathophysiological approach to treating diabetes advocates selecting treatment options that target the pathophysiologic defects associated with type 2 diabetes.2 Beta cell loss over time blunts the response to certain antidiabetic therapies and should be considered before choosing agents for intensification. Traditional treatment strategies for the management of type 2 diabetes modify treatment regimens only when specific glycemic targets are not met. This “failure-based” approach often results in a delay in advancing therapy.9 Landmark diabetes studies have predicted that early glycemic control can reduce the risk of development and progression of diabetic complications compared to late intensification.
References
- Weir GC, Bonner-Weir S. Islet β cell mass in diabetes and how it relates to function, birth, and death. Ann N Y Acad Sci. 2013;1281:92-105.
- DeFronzo RA, Eldor R, Abdul-Ghani M. Pathophysiologic approach to therapy in patients with newly diagnosed type 2 diabetes. Diabetes Care. 2013;36(suppl 2):S127-S138.
- Butler AE, Janson J, Bonner-Weir S, et al. β-cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes. 2003;52:102-110.
- Drucker DJ, Nauck MA. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet. 2006;368:1696-705.
- Meloni AR, DeYoung MB, Lowe C, et al. GLP-1 receptor activated insulin secretion from pancreatic β-cells: Mechanism and glucose dependence. Diabetes Obes Metab. 2013;15:15-27.
- Gerich JE. Role of the kidney in normal glucose homeostasis and in the hyperglycaemia of diabetes mellitus: Therapeutic implications. Diabet Med. 2010;27:136-142.
- Goldenberg RM, Steen O. Semaglutide: Review and place in therapy for adults with type 2 diabetes. Can J Diabet. 2019;43:136-145.
- Drucker DJ. Enhancing incretin action for the treatment of type 2 diabetes. Diabetes Care. 2003;26:2929-2940.
- Kendall DM, Cuddihy RM, Bergenstal RM. Clinical application of incretin-based therapy: Therapeutic potential, patient selection and clinical use. Am J Med. 2009;122:S37-S50.













